
Chemical applicability of Sombor indices Survey
Article Sidebar
Main Article Content
Abstract
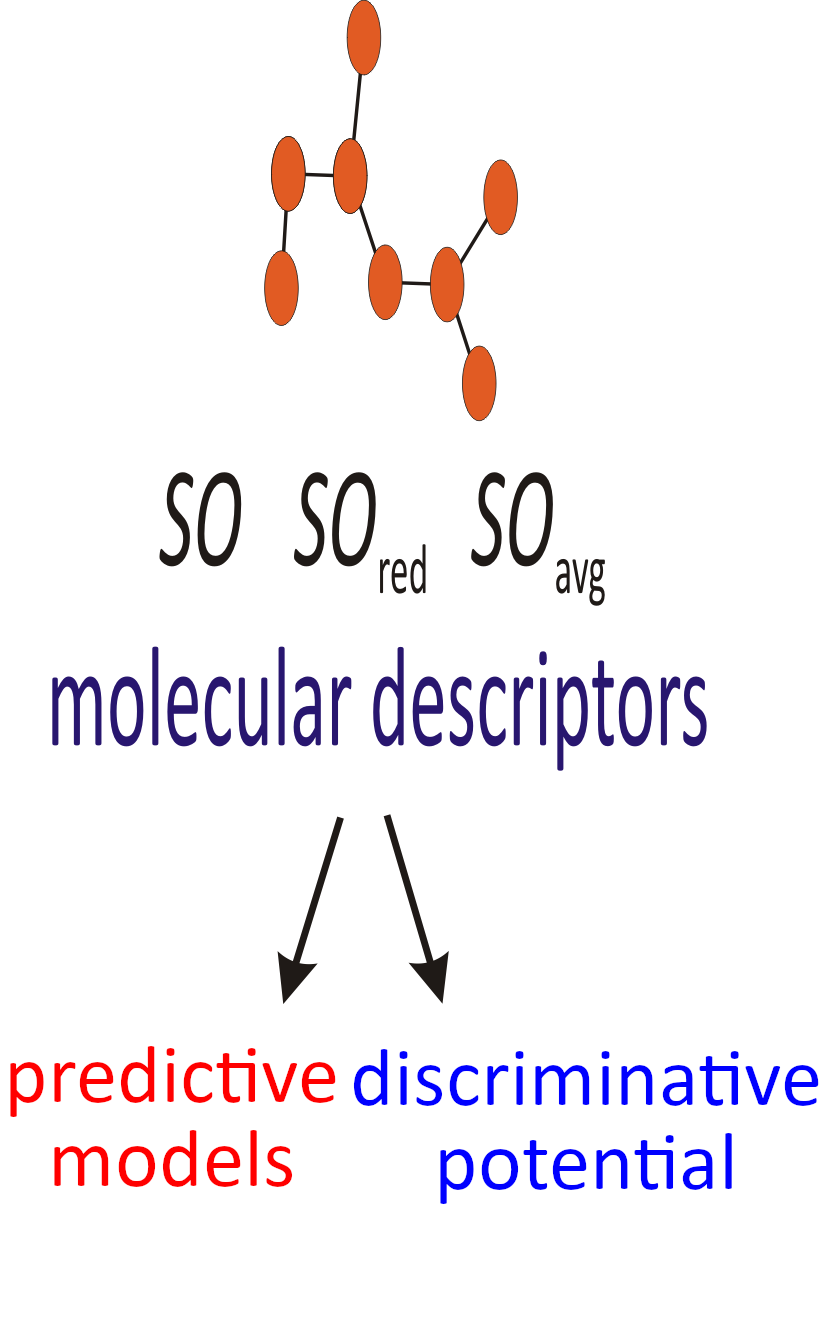
Recently, a novel class of degree-based topological molecular descriptors was proposed, the so-called Sombor indices. Within this study, the predictive and discriminative potentials of the Sombor index, the reduced Sombor index, and the average Sombor index were examined. All three topological molecular descriptors showed good predictive potential. The statistical data indicate that the reduced Sombor index preforms with a slightly better predictive potential. An external validation confirmed this finding. It was found that these degree-based indices exert modest discriminative potential, when tested on a large group of isomers.
Downloads
Metrics
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.

Authors retain copyright and grant the journal right of first publication with the work simultaneously licensed under a Creative Commons Attribution license 4.0 that allows others to share the work with an acknowledgement of the work's authorship and initial publication in this journal.




References
X. Ling, J. Bajorath, Comb. Chem. High Throughput Screen. 3 (2000) 363 (https://doi.org/10.2174/1386207003331454)
R. Todeschini, V. Consonni, Molecular Descriptors for Chemoinformatics, Wiley, Weinheim, 2009 (https://doi.org/10.1002/9783527628766)
H. Hong, Q. Xie, W. Ge, F. Qian, H. Fang, L. Shi, Z. Su, R. Perkins, W. Tong, J. Chem. Inf. Model. 48 (2008) 1337 (https://doi.org/10.1021/ci800038f)
S. Sahoo, C. Adhikari, M. Kuanar, B. K. Mishra, Curr. Comput. Aided Drug Des. 12 (2016) 181 (https://doi.org/10.2174/1573409912666160525112114)
A. R. Katritzky, E. V. Gordeeva, J. Chem. Inf. Comput. Sci. 33 (1993) 835 (https://doi.org/10.1021/ci00016a005)
M. Karelson, Molecular Descriptors in QSAR/QSPR, Interscience, New York, 2000 (ISBN: 978-0-471-35168-9)
M. Dehmer, K. Varmuza, D. Bonchev, Statistical Modelling of Molecular Descriptors in QSAR/QSPR, John Wiley & Sons, Weinheim, 2012 (https://doi.org/10.1002/9783527645121)
K. Roу, S. Kar, R. N. Das, A Primer on QSAR/QSPR Modeling-Fundamental Concepts, Springer, Cham, 2015 (https://doi.org/10.1007/978-3-319-17281-1)
J. Bajorath, J. Chem. Inf. Comput. Sci. 41 (2001) 233 (https://doi.org/10.1021/ci0001482)
F. Grisoni, D. Reker, P. Schneider, L. Friedrich, V. Consonni, R. Todeschini, A. Koeberle, O. Werz, G. Schneider, Mol. Inform. 36 (2017) 1600091 (https://doi.org/10.1002/minf.201600091)
N. Trinajstić, Chemical Graph Theory, Routledge, New York, 2018 (https://doi.org/10.1201/9781315139111)
D. Bonchev, Chemical Graph Theory: Introduction and Fundamentals, CRC Press, Boca Raton, FL, 1991 (ISBN: 978-0856264542)
I. Gutman, Selected Theorems in Chemical Graph Theory, University of Kragujevac, Kragujevac, 2017 (ISBN: 978-86-6009-039-5)
E. Estrada, G. Patlewicz, E. Uriarte, Indian J. Chem., A 42 (2003) 1315 (http://hdl.handle.net/123456789/20663)
R. Zanni, M. Galvez-Llompart, R. Garcia-Domenech, J. Galvez, Expert Opin. Drug Discov. 10 (2015) 945 (https://doi.org/10.1517/17460441.2015.1062751)
O. Ivanciuc, Curr. Comput. Aided Drug Des. 9 (2013) 153 (https://doi.org/10.2174/1573409911309020002)
I. Redžepović, B. Furtula, J. Comput. Aided Mol. Des. 34 (2020) 975 (https://doi.org/10.1007/s10822-020-00320-2)
I. Redžepović, Y. Mao, Z. Wang, B. Furtula, Int. J. Quantum Chem. 120 (2020) e26209 (https://doi.org/10.1002/qua.26209)
I. Redžepović, B. Furtula, J. Serb. Soc. Comput. Mech. Special Issue (2020) 37 (https://doi.org/10.24874/jsscm.2020.01.04)
R. Gozalbes, J. P. Doucet, F. Derouin, Curr. Drug Targets Infect. Disord. 2 (2002) 93 (https://doi.org/10.2174/1568005024605909)
I. Gutman, N. Trinajstić, Chem. Phys. Lett. 17 (1972) 535 (https://doi.org/10.1016/0009-2614(72)85099-1)
S. Nikolić, G. Kovačević, A. Miličević, N. Trinajstić, Croat. Chem. Acta 76 (2003) 113 (https://hrcak.srce.hr/103086)
I. Gutman, K. C. Das, MATCH Commun. Math. Comput. Chem. 50 (2004) 83 (http://match.pmf.kg.ac.rs/electronic_versions/Match50/match50_83-92.pdf)
M. Eliasi, A. Iranmanesh, I. Gutman, MATCH Commun. Math. Comput. Chem. 68 (2012) 217 (http://match.pmf.kg.ac.rs/electronic_versions/Match68/n1/match68n1_217-230.pdf)
A. Ali, N. Trinajstić, Mol. Inform. 37 (2018) 1800008 (https://doi.org/10.1002/minf.201800008)
Z. Raza, A. Ali, Int. J. Quantum Chem. 120 (2020) e26333 (https://doi.org/10.1002/qua.26333)
T. Došlić, B. Furtula, A. Graovac, I. Gutman, S. Moradi, Z. Yarahmadi, MATCH Commun. Math. Comput. Chem. 66 (2011) 613 (http://match.pmf.kg.ac.rs/electronic_versions/Match66/n2/match66n2_613-626.pdf)
I. Gutman, MATCH Commun. Math. Comput. Chem. 86 (2021) 11 (http://match.pmf.kg.ac.rs/content86n1.htm)
M. Randić, J. Math. Chem. 7 (1991) 155 (https://doi.org/10.1007/BF01200821)
www.moleculardecriptors.eu (accessed February 15, 2020)
F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot, E. Duchesnay, J. Mach. Learn. Res. 12 (2011) 2825 (https://www.jmlr.org/papers/volume12/pedregosa11a/pedregosa11a.pdf)
E. V. Konstantinova, J. Chem. Inf. Comput. Sci. 36 (1996) 54 (https://doi.org/10.1021/ci9502461)
M. Randić, J. Am. Chem. Soc. 97 (1975) 6609 (https://doi.org/10.1021/ja00856a001)
D. Vukičević, M. Gašperov, Croat. Chem. Acta 83 (2010) 243 (https://hrcak.srce.hr/62202).